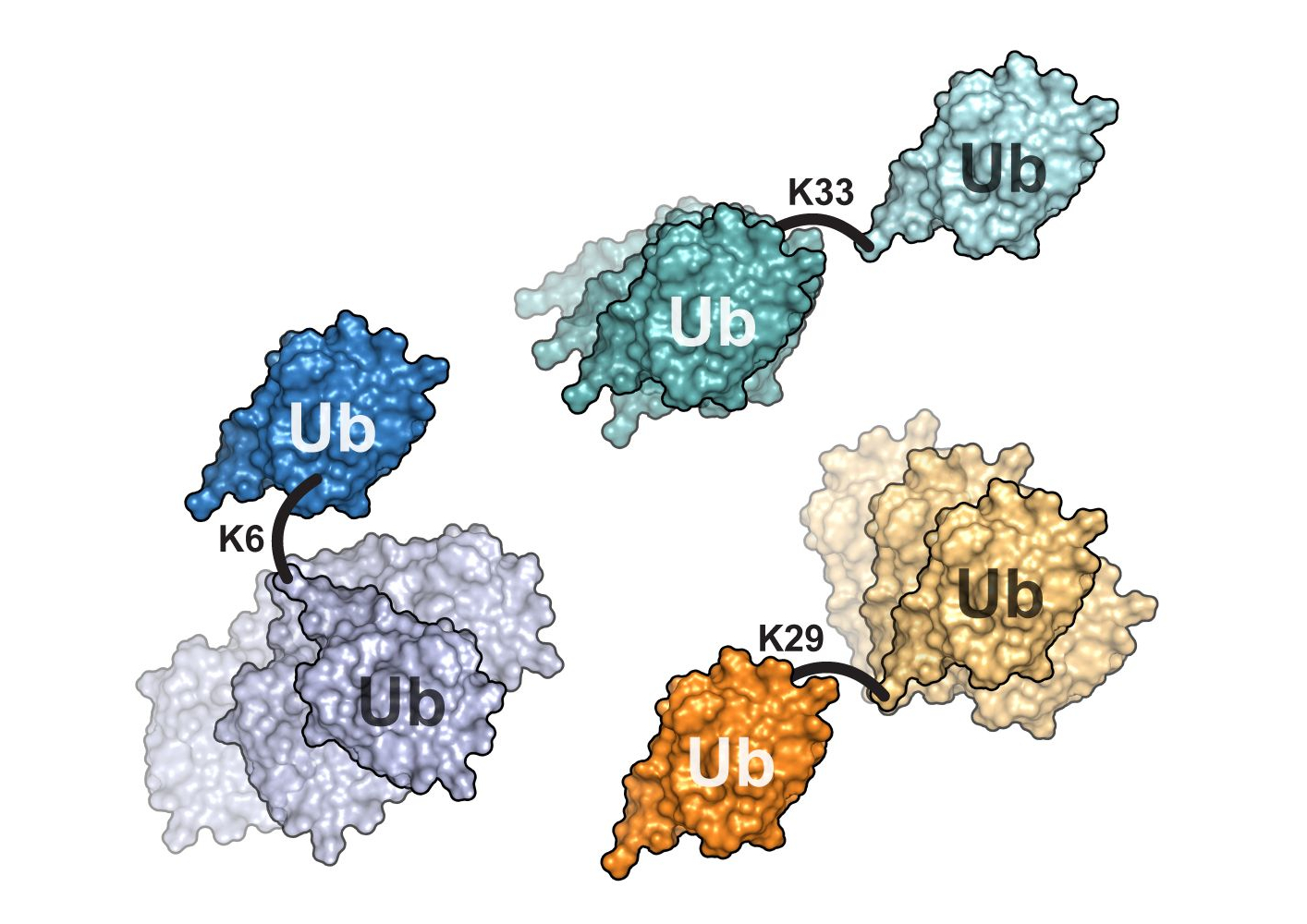
Deciphering the molecular dynamics of complex proteins
Which structures do complex proteins adopt in solution? Konstanz biophysicists answer this question using the example of ubiquitin dimers as well as a new combination of high-resolution NMR spectroscopy and sophisticated computer simulations.
Proteins consist of amino acids, which are linked to form long amino acid chains as specified by our genetic material. In our cells, these chains are not simply rolled up like strings of pearls, but fold into complex, three-dimensional structures. How a protein is folded decisively influences its function: It determines, for example, which other molecules a protein can interact with in the cell. Knowledge of the three-dimensional structure of proteins is therefore of great interest to the life sciences and plays a role in drug development, among other things.
"Unfortunately, elucidating the structure of a protein is anything but trivial, and focusing on a single state does not always provide meaningful information, especially if the protein is highly flexible in terms of its structure", says Tobias Schneider, a member of Michael Kovermann's lab team in the Department of Chemistry at the University of Konstanz. The reason: complex proteins often fold into several compact subunits, called domains, which in turn may be connected by flexible linkers. The more flexibly connected subunits are present, the more different three-dimensional structures a protein can theoretically adopt. "This means that a protein in solution, for example inside our cells, has several equal states and constantly switches between them", Schneider explains.
Tracking down the structural ensemble
A simple snapshot is not sufficient to fully describe the structural features of such multi-domain proteins, as it would capture only one of many states at a time. To get a detailed picture of the possible structures of such proteins, a smart combination of different methods is needed. In an article published in the journal Structure, Konstanz biophysicists led by Michael Kovermann and Christine Peter (also Department of Chemistry) present a corresponding approach using complementary methods.
"Through NMR spectroscopy, for example, we get information about the dynamic properties of such proteins. Complex computer simulations, on the other hand, provide a good overview of the range of possible folds", explains Kovermann. "So far, no general approach that comprehensively maps the dynamic and structural properties of multi-domain proteins had existed". The researchers from Konstanz therefore devised a workflow that combines NMR spectroscopy and computer simulations, allowing them to obtain information on both properties with high temporal and spatial resolution.
Proof of feasibility included
The researchers also provided evidence that the method works: They examined various ubiquitin dimers. These consist of two units of the protein ubiquitin that are linked by a flexible bond, just like the situation in cells. It is thus a prime example of a multi-domain protein for which different structural models have been suggested so far and which is of great scientific interest.
The researchers were able to show that the ubiquitin dimers they studied exhibit a high structural variability and that this can be described in detail using the developed combination of methods. The results also explain the different structural models that currently exist of ubiquitin dimers. "We are convinced that our approach – combining complementary methods – will work not only for ubiquitin dimers but also for other multi-domain proteins", Schneider says. "Our study opens new avenues to better understand the high structural diversity of these complex proteins that plays a crucial role in their biological functions".
Key facts:
- Original publication: T. Schneider, K. Sawade, F. Berner, C. Peter & M. Kovermann (2023) Specifying conformational heterogeneity of multi-domain proteins at atomic resolution. Structure; doi: https://doi.org/10.1016/j.str.2023.07.008
- Biophysicists from Konstanz develop method for detecting the conformational landscape of multidomain proteins with atomic resolution
- Workflow combines chemical biology, NMR spectroscopy and molecular dynamics simulations
- Funding: SFB969 of the German Research Foundation (DFG) and Young Scholar Fund of the University of Konstanz